Panoramica
L’ordinata serie di eventi che porta alla duplicazione di tutte le componenti cellulari e che culmina con la divisione di due cellule geneticamente identiche viene chiamata ciclo cellulare. Mentre una parte di tutte le cellule somatiche che costituisce il nostro corpo dà luogo a frequenti divisioni cellulari, il fenomeno della morte cellulare programmata elimina le cellule danneggiate. Sia il ciclo cellulare che la morte cellulare programmata sono fondamentali per il corretto sviluppo degli organismi pluricellulari e per il funzionamento di tessuti ed organi. Mentre questi due processi sono stati storicamente studiati separatamente l’uno dall’altro, il nostro gruppo di ricerca si ripropone di mappare i circuiti molecolari che consentono a questi due processi di comunicare tra loro. In particolare, siccome alterazioni di entrambi i processi preludono all’insorgenza di tumori, la comprensione dei meccanismi molecolari che consentono ai due processi di comunicare può contribuire a concepire nuove strategie terapeutiche per combattere il cancro.
Linee di ricerca
I meccanismi cellulari che rispondono alla mancata divisione
Il mancato completamento della divisione cellulare (o citochinesi) è uno dei difetti più comuni del ciclo cellulare e predispone le cellule somatiche alla trasformazione tumorale. Sorprendentemente però, alcuni tipi cellulari specifici nel nostro corpo, come gli epatociti e i cardiomiociti, perseguono attivamente la mancata divisione cellulare come parte del loro programma di differenziamento. Nonostante queste differenze tra tipi cellulari diversi, la mancata citochinesi riduce immancabilmente la tendenza a intraprendere un ciclo cellulare successivo. La nostra ricerca si ripropone di:
-
Comprendere che cosa consente alle cellule di riconoscere la mancata divisione cellulare
-
Quali sono i segnali cellulari che si generino e come questi cambino la fisiologia cellulare
Nei nostri studi dedichiamo molta attenzione al centrosoma, un organello che è normalmente presente in singola copia per cellula ma che a seguito della mancata divisione si presenta in copie multiple. Utilizziamo pertanto una combinazione di strumenti di ingegneria genetica, microscopia a fluorescenza e proteomica per comprendere il contributo dei centrosomi multipli nella trasmissione di segnali che modificano il comportamento cellulare.
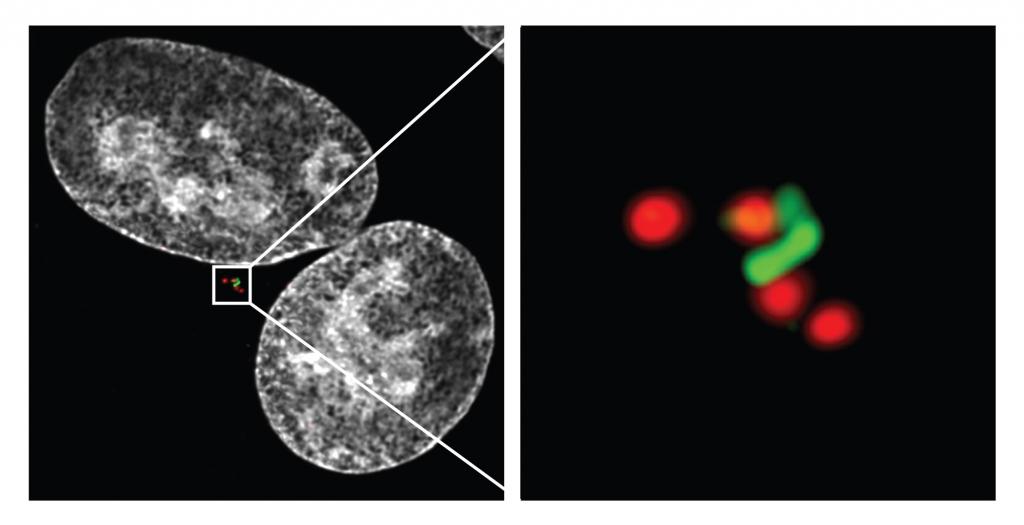 Microscopia confocale di una cellula di un carcinoma al polmone che ha mancato la divisione cellulare.
Microscopia confocale di una cellula di un carcinoma al polmone che ha mancato la divisione cellulare.
Sinistra: l’intera cellula. I due nuclei sono evidenziati in bianco mentre diverse proteine del centrosoma sono evidenziate in rosso e verde.
Destra: maggior ingrandimento dei centrosomi duplicati giustapposti.
Alla riscoperta di p53: funzioni non canoniche in risposta ad errori della divisione cellulare.
La proteina p53, il più noto e studiato “soppressore di tumori”, è stata studiata principalmente come fattore di trascrizione in risposta a danni al DNA genomico. Nonostante sia noto dagli anni 70 che p53 si attivi anche in risposta a difetti della divisione cellulare, si conosce sorprendentemente poco di come p53 modifichi il comportamento cellulare in risposta a mancata citochinesi o all’anormale segregazione di singoli cromosomi. Combinando tecniche di biochimica classica e di sequenziamento dell’RNA di ultima generazione ci riproponiamo di studiare il contributo di p53 a valle dei difetti mitotici. Così facendo ci aspettiamo di portare alla luce nuove sfaccettature dell’azione di p53 nel prevenire l’insorgenza dei tumori.
Membri del gruppo
- Luca Fava, PI
- Alessia Mattivi, laboratory manager
- Florian Bellutti, ricercatore post-doc
- Matteo Burigotto, ricercatore post-doc
- Iva Dzhilyanova, dottoranda
- Vincenza Vigorito, dottoranda
- Selene Tessadri, borsista predoc
- Stefano Li Veli, studente MSc
Collaborazioni in corso
- Andreas Villunger, Medical University of Innsbruck, Austria
- Andreas Strasser, WEHI, Australia
- Stefano Maffini & Andrea Musacchio, MPI Dortmund, Germany
Sponsor
- 2020-2025, AIRC
- 2019-2022, PRIN MIUR
- 2017-2022, Armenise-Harvard Career Development Award
Pubblicazioni selezionate
Ghetti G, Burigotto M, Mattivi A, Magnani G, Casini A, Bianchi A, Cereseto A, Fava LL* (2021) CRISPR/Cas9 ribonucleoprotein-mediated knockin generation in hTERT-RPE1 cells. STAR Protocols. 2, 100407
Burigotto M, Mattivi A, Migliorati D, Magnani G, Valentini C, Roccuzzo M, Offterdinger M, Pizzato M, Schmidt A, Villunger A, Maffini S, Fava LL* (2021) Centriolar distal appendages activate the centrosome-PIDDosome-p53 signalling axis via ANKRD26. The EMBO journal. 40, e104844
Sladky VC, Knapp K, …, Fava LL, …, Alain de Bruin & Andreas Villunger (2020) E2F-Family Members Engage the PIDDosome to Limit Hepatocyte Ploidy in Liver Development and Regeneration. Developmental Cell 52, 1-15
Liccardi G, Ramos Garcia L, …, Fava LL, …, Bianchi K & Meier P (2019) RIPK1 and Caspase-8 Ensure Chromosome Stability Independently of Their Role in Cell Death and Inflammation. Molecular Cell 73, 413-428
Fava LL*, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA & Villunger A* (2017) The PIDDosome activates p53 in response to supernumerary centrosomes. Genes & development 31, 34–45
Haschka MD, Soratroi C, Kirschnek S, Hacker G, Hilbe R, Geley S, Villunger A* & Fava LL* (2015) The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nature communications 6, 6891
*=autori corrispondenti