Overview
Work in our laboratory covers a broad spectrum of topics related to protein dynamics across physiology and disease. We investigate these phenomena by integrating various computational, chemical, biophysical, biochemical and cellular technologies, aiming to capitalize on such information to develop new experimental therapies for neurodegenerative diseases.
Research directions
Protein expression and function in eukaryotic cells are tightly harmonized processes modulated by the combination of different layers of regulation, including transcription, processing, stability, and translation of messenger RNA, as well as assembly, maturation, sorting, recycling, and degradation of polypeptides. Integrating all these pathways and the protein quality control machinery, deputed to avoid the production and accumulation of aberrantly folded proteins, determines protein homeostasis. Over the last decade, the combined development of accurate time-resolved experimental techniques and efficient computer simulations has opened the possibility of investigating biological mechanisms at atomic resolution with physics-based models. A meaningful example is the reconstruction of protein folding pathways at atomic resolution, which has enabled the characterization of the folding kinetics of biologically relevant globular proteins. Combining these innovative computational technologies with rigorous experimental approaches reveals the existence of non-native metastable states transiently appearing along the folding process of such proteins. We are primarily interested in testing the possibility that such protein folding intermediates could play roles in disparate biological processes, from the post-translational regulation of protein expression to disease-relevant protein misfolding mechanisms. We aim to exploit the information encoded into protein folding pathways to design an entirely new generation of pharmacological agents capable of promoting the selective degradation of protein targets.
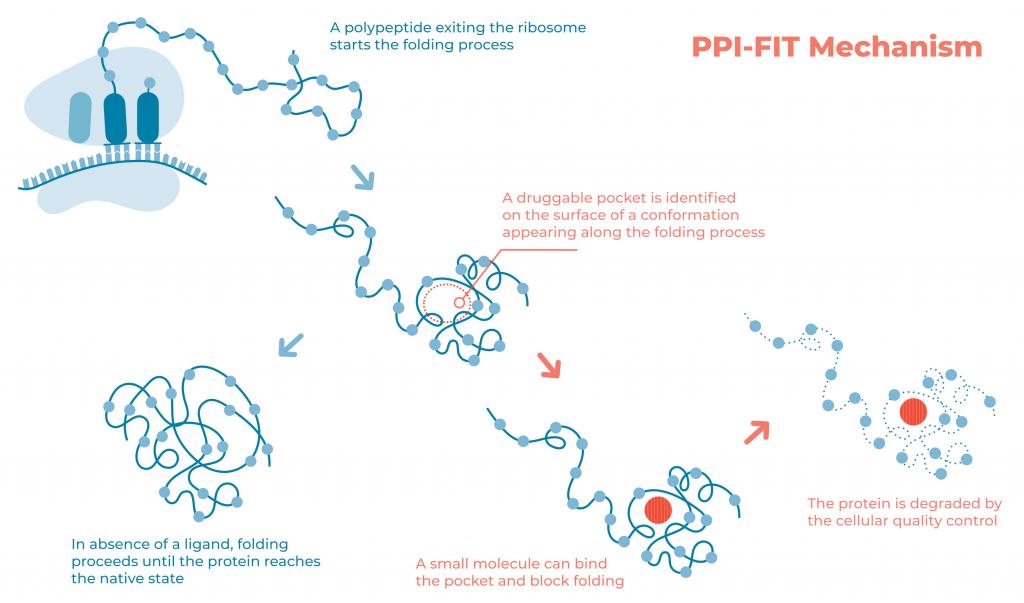
The image illustrates the rationale underlying the PPI-FIT (Pharmacological Protein Inactivation by Folding Intermediate Targeting) paradigm. Polypeptides exiting the ribosomal channel start the folding process. Folding intermediates transiently appearing along this process could be targeted with small molecules binding to unique pockets on their surface. Such a binding event ultimately interferes with the protein folding process, prompting the quality control machinery of the cells to reroute the polypeptide to degradation pathways. The technology is currently exploited by Sibylla Biotech (www.sibyllabiotech.it).

Patch of the Zeprion2023 space mission. The experiment aims at performing the crystallization of a protein folding intermediate of the prion protein bound to a ligand on the International Space Station. The mission builts on the hypothesis that the absence of convective motion, as obtained in microgravity conditions, could prevent protein aggregation and favor crystallization. The experiment is made possible by the collaboration with the Israeli company SpacePharma (https://www.spacepharma.health/) and its Lab-On-A-Chip proprietary system.
JOIN OUR TEAM!
If you're a student at the bachelor, master or doctoral level with a strong interest in protein biology and experimental drug discovery, then our group might be the perfect fit for you. Protein dynamics is a complex process, and tackling it requires a broad range of scientific approaches. In our laboratory, you will have the opportunity to work with a team of researchers from highly diverse scientific backgrounds, gaining exposure to fields like computational biophysics, theoretical and synthetic chemistry, biochemistry and cell biology. By training with us, you will develop a deep understanding of the molecular mechanisms involved in protein folding and misfolding and have the opportunity to use advanced techniques to probe this process and discover new drugs for neurological disorders. By joining us, you will also be part of a vibrant community of scientists who are passionate about their work. You'll have the opportunity to participate in regular research meetings, conferences, and seminars, which will help you develop a broad network of colleagues and collaborators.
Group members
- Emiliano Biasini, Associate Professor
- Marta Rigoli, Postdoctoral Fellow
- Valerio Bonaldo, PhD student
- Ilaria Zeni, PhD student
- Nicole Innocenti, PhD student
- Dino Gasparotto, PhD Student
Collaborations
- Ines Mancini, Graziano Lolli, Alberto Inga & Yari Ciribilli, University of Trento, ITALY
- Pietro Faccioli, University of Milano-Bicocca, ITALY
- Maria Letizia Barreca, Giuseppe Manfroni & Francesca Fallarino, University of Perugia, ITALY
- Jesús R. Requena, University of Santiago de Compostela, SPAIN
- Romolo Nonno, Istituto Superiore di Sanità, ITALY
- Pietro Roversi, CNR, ITALY
- Giovanni Nardo & Valentina Bonetto, Mario Negri Institute in Milan, ITALY
- Alan Ianeselli, Free University of Bozen, ITALY
- SpacePharma r&d, ISRAEL
- Sibylla Biotech S.p.A., ITALY
Funding
Bando: PRIN 2022 (D.D. 104/22)
Non-canonical small molecule degraders of the prion protein to treat neurodegenerative disorders
Emiliano Biasini, Responsabile di Unità
Codice Protocollo: 2022PP8WNZ CUP: E53D23009460006

Selected publications
Biasini E, Faccioli P. Functional, Pathogenic, and Pharmacological Roles of Protein Folding Intermediates. Proteins. 2023 Feb 13. doi: 10.1002/prot.26479.
https://pubmed.ncbi.nlm.nih.gov/36779817
Spagnolli G, Massignan T, et al. Pharmacological inactivation of the prion protein by targeting a folding intermediate. Commun Biol. 2021 Jan 12;4(1):62. doi: 10.1038/s42003-020-01585-x.
https://pubmed.ncbi.nlm.nih.gov/33437023
Manni G, Lewis V, Senesi M, Spagnolli G, Fallarino F, Collins SJ, Mouillet-Richard S, Biasini E. The cellular prion protein beyond prion diseases. Swiss Med Wkly. 2020 Apr 24;150:w20222. doi: 10.4414/smw.2020.20222.
https://pubmed.ncbi.nlm.nih.gov/32330284
Spagnolli G, Rigoli M, Orioli S, Sevillano AM, Faccioli P, Wille H, Biasini E & Requena JR. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019 Jul 11;15(7):e1007864. doi: 10.1371/journal.ppat.1007864.
https://pubmed.ncbi.nlm.nih.gov/31295325
Rigoli M, Spagnolli G, Faccioli P, Requena JR & Biasini E. Ok Google, how could I design therapeutics against prion diseases? Curr Opin Pharmacol. 2019 May 3;44:39-45. doi: 10.1016/j.coph.2019.03.015.
https://pubmed.ncbi.nlm.nih.gov/31059982
Biasini E. A designer chaperone against prion diseases. Nat Biomed Eng. 2019 Mar;3(3):167-168. doi: 10.1038/s41551-019-0367-6.
https://pubmed.ncbi.nlm.nih.gov/30948815
For a complete list: https://www.ncbi.nlm.nih.gov/pubmed/?term=emiliano+biasini