Overview
The ordered series of events leading to the duplication of all cellular components, culminating in the generation of two genetically identical daughter cells via cell division, is known as the cell division cycle. While a fraction of the cells within multicellular organisms divides constantly, the process of genetically programmed cell death (e.g. apoptosis) depletes non-functional or damaged cells from the body. Both the cell cycle and cell death are instrumental for organism to develop and maintain tissue homeostasis. For many years, anecdotal evidence suggested that both processes are interlinked. Our group aims to unravel the molecular circuitry that allows the cross-communication between the cell cycle and cell death machineries. Importantly, as a deregulation of both processes has been causally linked to cancer, we hope that by providing a better understanding of those connections we will contribute to the design of innovative approaches to combat cancer.
Research directions
-
A sensing mechanism to monitor duplication of the cellular components
Failure in the physical separation of two cells at the end of cell division, i.e. cytokinesis, is one of the most common malfunctions of the cell division cycle, predisposing cells to neoplastic transformation. Strikingly, however, specific cell types in the human body, such as hepatocytes and cardiomyocytes, actively implement cytokinesis failure as part of a developmental program to duplicate their cellular components. Despite this heterogeneity, cytokinesis failure is invariably followed by a reduction in the propensity of cells to commit to additional cell cycles. What enables cells to recognize the occurrence of cytokinesis failure and to restrain their proliferative potential? Centrosome abundance appears crucial for determining the cellular behaviour in response to cytokinesis failure. Centrosomes, the main microtubule organizing centres in the cell, duplicate once per cell cycle, allowing a tight regulation of their numbers. Using a combination of genetic tools, advanced live cell imaging and proteomics, we wish to uncover the mechanisms that allow cells to sense cytokinesis failure, with special attention on the role of supernumerary centrosomes.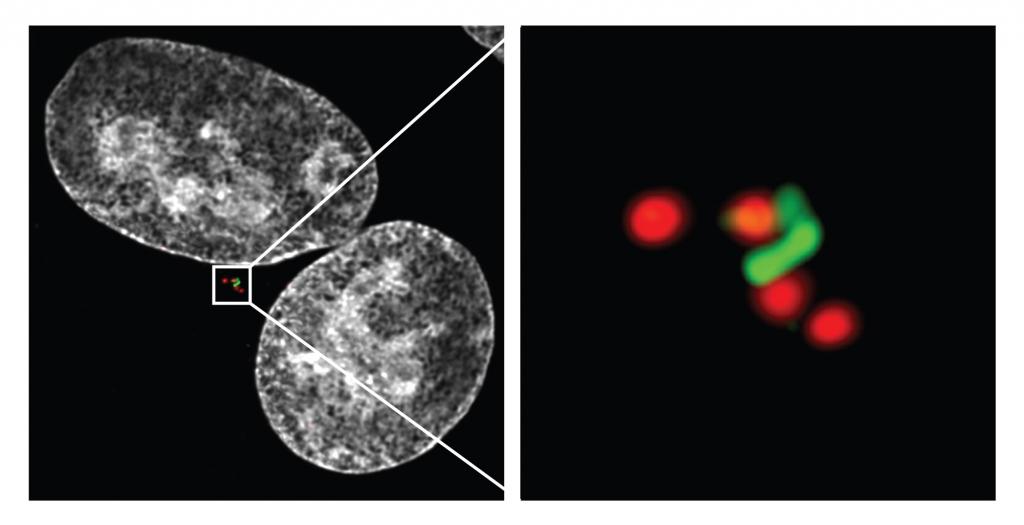
Immunofluorescence staining of a lung adenocarcinoma cell that failed cytokinesis
Left: overview of the whole cell: nuclei are stained white, different centrosomal markers are stained red and green.
Right: blow-up of the duplicated centrosomes. -
Re-invented p53: non-canonical functions in response to cell division errors
The protein p53, one of the best-known tumour suppressors, has been studied most intensively as a transcription factor responding to acute damage of the DNA. Though cytokinesis failure has been known to activate p53 since the 1970s, surprisingly little is understood about how p53 influences cell fate in response to cell division errors such as cytokinesis failure or the mis-segregation of individual chromosomes. By combining classic biochemistry techniques and next-generation RNA sequencing, we aim to systematically dissect p53 activation in response to cell division errors. This project will reveal a hitherto underappreciated role of p53 in tumour suppression.
Group members
- Luca Fava, PI
- Alessia Mattivi, laboratory manager
- Florian Bellutti, postdoctoral fellow
- Matteo Burigotto, postdoctoral fellow
- Iva Dzhilyanova, PhD student
- Vincenza Vigorito, PhD student
- Selene Tessadri, predoc fellow
- Stefano Li Veli, MSc student
On-going Collaborations
- Andreas Villunger, Medical University of Innsbruck, Austria
- Andreas Strasser, WEHI, Australia
- Stefano Maffini & Andrea Musacchio, MPI Dortmund, Germany
Funding
- 2020-2025, AIRC
- 2019-2022, PRIN MIUR
- 2017-2022, Armenise-Harvard Career Development Award
-
Bando: PRIN 2022 (D.D. 104/22)
Targeting mitotic catastrophe to revert drug resistance in acute myeloid leukemia: a systems-based approach
Luca Fava, Responsabile di Unità
Codice Protocollo: 2022L8RAKN CUP: E53D23004860006
Selected publications
Ghetti G, Burigotto M, Mattivi A, Magnani G, Casini A, Bianchi A, Cereseto A, Fava LL* (2021) CRISPR/Cas9 ribonucleoprotein-mediated knockin generation in hTERT-RPE1 cells. STAR Protocols. 2, 100407
Burigotto M, Mattivi A, Migliorati D, Magnani G, Valentini C, Roccuzzo M, Offterdinger M, Pizzato M, Schmidt A, Villunger A, Maffini S, Fava LL* (2021) Centriolar distal appendages activate the centrosome-PIDDosome-p53 signalling axis via ANKRD26. The EMBO journal. 40, e104844
Sladky VC, Knapp K, …, Fava LL, …, Alain de Bruin & Andreas Villunger (2020) E2F-Family Members Engage the PIDDosome to Limit Hepatocyte Ploidy in Liver Development and Regeneration. Developmental Cell 52, 1-15
Liccardi G, Ramos Garcia L, …, Fava LL, …, Bianchi K & Meier P (2019) RIPK1 and Caspase-8 Ensure Chromosome Stability Independently of Their Role in Cell Death and Inflammation. Molecular Cell 73, 413-428
Fava LL*, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA & Villunger A* (2017) The PIDDosome activates p53 in response to supernumerary centrosomes. Genes & development 31, 34–45
Haschka MD, Soratroi C, Kirschnek S, Hacker G, Hilbe R, Geley S, Villunger A* & Fava LL* (2015) The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nature communications 6, 6891
*=corresponding authors
For a complete list see: https://www.ncbi.nlm.nih.gov/pubmed/?term=fava+ll